Abstract
From potentiodynamic and potentiostatic electrochemical methods it was assessed, for the first time, the kinetics and mechanism involved during the electrochemical synthesis of palladium nanoparticles, PdNPs, from a deep eutectic solvent, DES, formed by the choline chloride and ethylene glycol eutectic mixture at 298 K. It was found that the potentiostatically electrodeposited PdNPs were formed through multiple 3D nucleation with diffusion-controlled growth. The nucleation frequency and the nucleation active sites increased linearly and exponentially with applied potential, respectively, and the Pd(II) ions diffusion coefficient, D, in DES was D = (2.77 ± 0.19) 10−7 cm2s−1. The morphology and composition of the PdNPs were characterized by SEM and XPS, respectively. The PdNPs were homogeneously dispersed onto the GCE surface, with monodispersed size ((41 ± 5) nm) and were formed mainly by metallic palladium with small amounts of PdO and PdO2. Furthermore, it was also shown that the GCE-PdNPs modified electrode depicts a high catalytic activity toward formic acid electrochemical oxidation reaction, revealing a mass activity of (4200 ± 100) mA mgPd−1 at the peak potential, much greater than those reported so far for other PdNPs synthesized by means of sophisticated, time-consuming methods.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Palladium nanoparticles (PdNPs), have been reported to be useful for several important applications, namely, as a highly sensitive electrochemical sensor for methanol determination,1 catalytic oxygen reduction reaction in glucose hybrid biofuel cells,2 catalytic oxidation of hydrazine in a hybrid self-powered N2H4 sensor,3 for induced cancer cell death,4 for hydrogen storage5,6 and sensing7,8 and in particular as anode materials for formic acid electrooxidation, in direct formic acid fuel cells (DFAFC).9–26 In order to synthesize the PdNPs, both chemical and electrochemical methods have been reported.27 The chemical methods9–20 are generally time-consuming multi-step processes that require the use of both reductive agents (i.e. sodium citrate or sodium borohydride) and stabilizing molecules (including organic ligands, salts/surfactants, polymers and dendrimers). In contrast, the electrochemical methods21–26,28–31 allow the nanostructures growth through only one step, directly onto the final support, without needing reductive and stabilizing agents, or further sample preparation, thus improving the electron pathway within the substrate, nanostructure, and environment.32–37 On consideration of the aforementioned facts and to the best of our knowledge, this work presents for the first time an in-depth characterization of the electrochemical synthesis of PdNPs onto a glassy carbon electrode (GCE) from a deep eutectic solvent formed by the mixture of choline chloride and ethylene glycol at room temperature. Moreover, the resulting modified electrode (GCE/PdNPs) was used for the electrochemical oxidation of formic acid, which is commonly used in direct alcohol fuel cells. Notwithstanding, it is important to mention that several aspects related with the Pd electrodeposition from DES have already been reported, for instance: the Particle/Solvent interactions of Pd Nanoparticles electrodeposited from a 2:1 urea:choline Cl− DES,31 the effect of additives and current mode on surface morphology of Pd films from a non-aqueous deep eutectic solution38 and the electrochemical growth of mesoporous metallic thin films by anodic dissolution in deep eutectic solvents.39
Experimental
The deep eutectic solvent was prepared by mixing choline chloride and ethylene glycol in a 1:2 molar ratio at 298 K, through magnetic stirring for 12 hours until achieving a transparent homogenous liquid. The previously dried PdCl2 salt was dissolved in the DES under stirring for 12 hours, thus becoming the electrolyte solution. The water content of the electrolyte solution was determined by Karl Fischer coulometric titration using a Titrino Coulometer model 756 from Metrohm giving less than 0.1%. All reagents were analytical grade from Sigma-Aldrich.
Electrochemical study
A three-electrode electrochemical cell was used (100 mL capacity) fitted with a Lauda RMS Circulator with RM6 Refrigerating Water Bath Chiller, to set the cell temperature at 298 ± 0.24 K. The working electrode was a glassy carbon electrode (GCE), with 0.1963 cm2 exposed area; the counter electrode was a platinum wire and a silver wire was used as pseudo reference electrode (Ag QRE), to which all potentials were quoted. The palladium nucleation and growth mechanism onto a GCE from the DES, was explored through electrochemical tests such as cyclic voltammetry (CV) and chronoamperometry (CA). The GCE was polished with diamond spray down to 0.25 μm, sonicated in methanol for 30 minutes, and finally rinsed with acetone to remove any residual particles and improve the surface adherence of the electrodeposited metal. CV and CA tests were carried out in an AutoLab PGSTAT100 potentiostat-galvanostat coupled to a PC running the NOVA 2.1.3 software for data acquisition and experimental control. These tests were carried out for both pure DES and Pd(II)/DES solutions.
SEM analysis
Morphology of palladium deposited on the GCE from DES was obtained by means of Scanning Electron Microscopy (SEM) using a model Nova-2000 Nanolab double beam FEI SEM instrument with secondary electrons detector.
XPS analysis
The deposit composition was determined by X-ray photoelectron spectroscopy (XPS) measurements, performed in an Escalab 250 Thermo Scientific (base pressure ∼2 × 10−9 mbar) equipment with an Al filament emitting X rays at 1486.6 eV. The Avantage 5 software was used for fitting the XPS profiles with Gaussian-Lorentzian (GL) line shape functions, calibrated by assigning a binding energy (BE) value of 285 eV to the C1s.
Results and Discussion
Potentiodynamic study
Figure 1 shows a typical CV recorded on the GCE working electrode in choline chloride-ethylene glycol eutectic mixture containing 5 mM PdCl2 at 298 K. A well resolved peak clearly formed during the cathodic scan, associated with the reaction Pd(II)DES + 2e− = Pd(s), displaying a current density, jcp, that increased linearly with the square root of the potential scan rate, v, as shown in the inset in Figure 1, as described by the Berzins-Delahay equation for the reversible and diffusion-controlled deposition of metals onto solid electrodes,40 which for 298 K corresponds to Eq. 1.
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0001.gif)
Where z is the number of electrons trasferred during Pd nucleation, C0 (mol L−1) and D (cm2s−1) are the bulk concentration and Pd(II) diffusion coefficient, respectively.
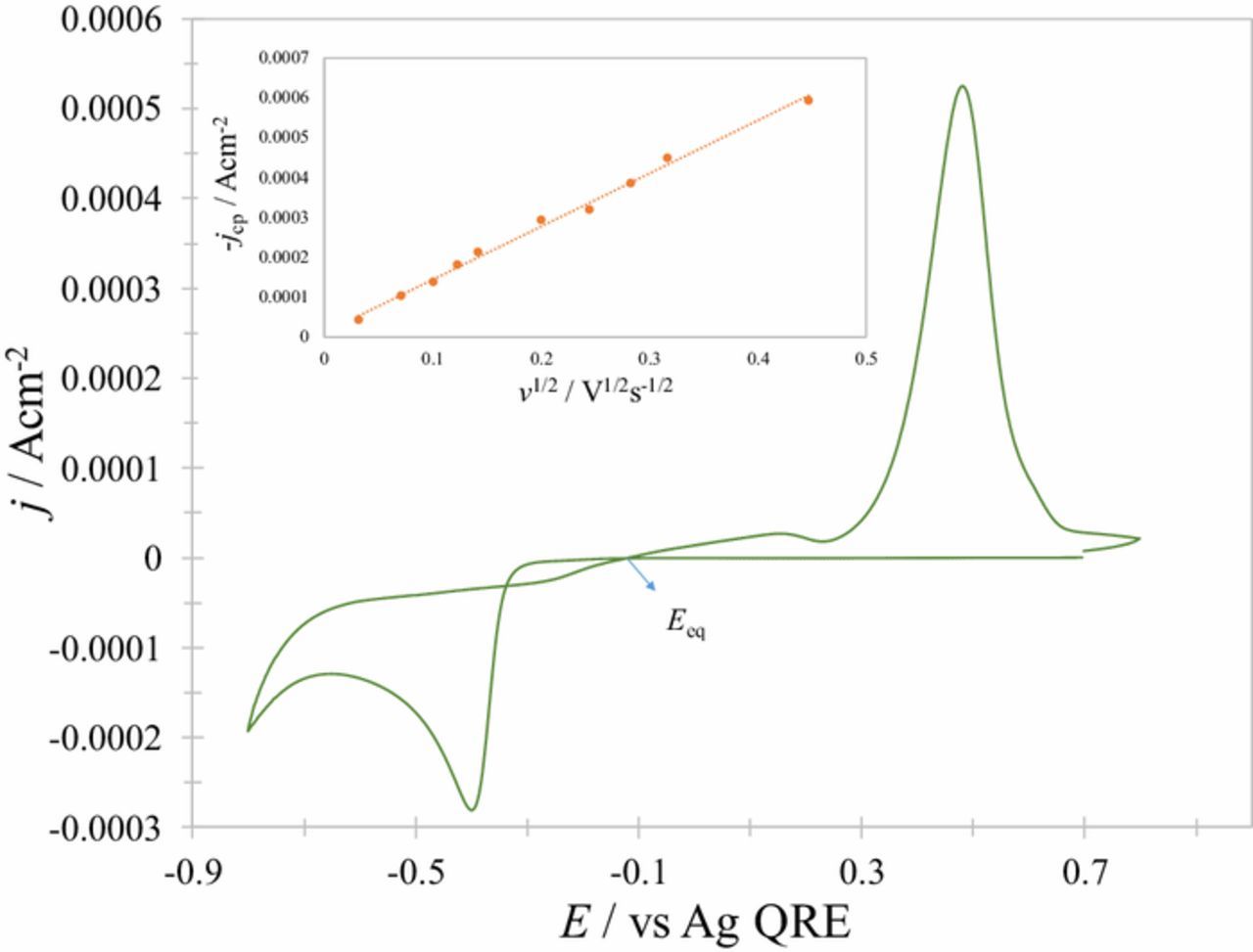
Figure 1. Typical experimental CV recorded on GCE working electrode in choline chloride:ethylene glycol eutectic mixture containing 5 mM PdCl2, starting the potential scan at 0.7 V in the negative direction at 40 mVs−1 sweep rate at 298 K. The inset shows the dependence of the cathodic peak current density, jcp, against the square root of the potential scan rate, v. The equation: jcp(Acm− 2) = 0.002(Acm− 2/V1/2s− 1/2) v1/2 was linearly fitted in the experimental data, thus giving the dashed line.
Furthermore, the CV in Figure 1 depicts a crossover between the cathodic and anodic voltammetric scans, characteristic of nucleation processes; therefore, the potentiodynamic analysis indicates that Pd deposition on the GCE occurs by a diffusion-controlled nucleation mechanism. On the reverse sweep of this CV, the palladium deposition occurs until the equilibrium potential, Eeq = −0.13 V, is reached ((second cross-over) on the CV at null current). After Eeq, the formation of a notable stripping peak, corresponds to the oxidation of the Pd deposited on the electrode. From the Eeq value and the applied potential, E, it is possible to calculate the nucleation overpotential, (η = E−Eeq).
Potentiostatic study
Figure 2 shows a family of potentiostatic current density transients recorded in the system GCE/5 mM PdCl2 in the choline chloride and ethylene glycol DES at different applied potentials within the cathodic peak zone shown in Figure 1. As previously described by Aldana-González et al.,33 it is possible to obtain solely the palladium electrochemical response in the DES by subtracting the experimental j-t plots recorded in DES, without Pd(II) ions (named jDES), related with pseudo-capacitive processes occurring during the double layer formed by the interface GCE/DES, from the plots obtained at the same conditions, but in the presence of Pd(II) ions (termed jPd-DES), see for instance the inset in Figure 2.
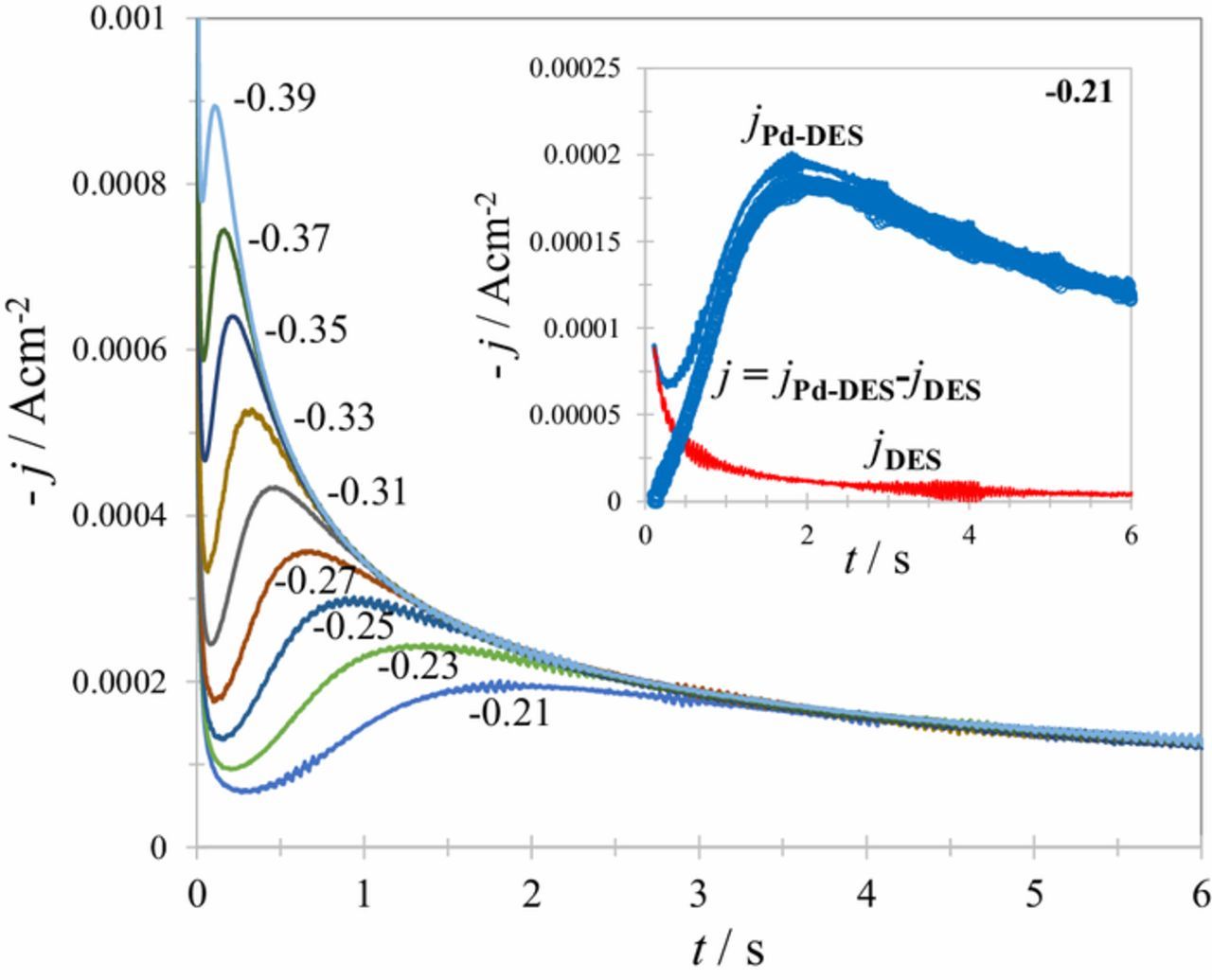
Figure 2. Family of experimental potentiostatic current density transients recorded in the system GCE/5 mM PdCl2, dissolved in the choline chloride and ethylene glycol DES at the different overpotentials indicated in the figure, in V. The inset shows the potentiostatic current density transient (markers) obtained at η = −0.21 V after subtraction of the respective blank (jDES) from the plot obtained at the same conditions, but in the presence of Pd(II) ions (jPd-DES).
Figure 3a shows the resulting family of current density transients after subtracting the respective blanks. It has been recently shown that the residual water in the DES can be simultaneously reduced on the growing surfaces of metallic centers, for instance during the Ni,33,41 Co35 and Cr34 electrochemical deposition on different substrates. As shown in these works, a useful criterion to assess the occurrence of concomitant reactions during the catalytic metal centers electrodepositon (like protons or water reduction)42,43 is to compare the experimental current density transients, normalized by means of the coordinate of their respective current density maxima (tm, jm), with the theoretical plots (j/jm)2 vs. t/tm for instantaneous and progressive nucleation mechanisms developed by Scharifker et al.44,45 If the experimental plots lie within the bounds of the theoretical limiting expressions, it is reasonable to assess that no concurrent faradaic simultaneous reactions are occurring on the surfaces of the growing metallic centers. According with this criterion, presently it can be noted from Figure 3b that there is no evidence to suggest the simultaneous reduction of residual water in the DES on the surfaces of PdNPs; thus, the experimental current transient will be analyzed with the theoretical framework, see Eq. 2, proposed by Heerman and Tarallo46 for the analysis of current density transients involving the electrochemical nucleation with diffusion-limited growth. The inset in Figure 3a depicts an example of the capacity of the Heerman and Tarallo model to describe the experimental current density transient recorded during the electrochemical synthesis of PdNPs from the DES, by means of nonlinear fitting of Eq. 8 into the experimental data. The best-fit parameters obtained are shown in Table I. From parameter P1, see Eq. 6, it was possible to calculate the Pd(II) ions diffusion coefficient in this DES as (2.77 ± 0.19) × 10−7 cm2s−1. It is important to mention that Katayama et al.,47 reported a value of 1.2 × 10−7 cm2 s−1 for the diffusion coefficient of the ion [PdCl4]2− dissolved in hydrophobic ionic liquid, 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide at 25°C using the Cottrell's equation while Lanzinger et al.,48 reported a value of 0.19 × 10−7 cm2 s−1 in the ChCl:EG eutectic mixture at 50°C using the Randles–Sevcik equation. Moreover, from Table I it can be noted that parameter P2, which is directly proportional to the number density of active sites, N0, increases exponentially with the applied potential, E, meanwhile the nucleation frequency, A, varies linearly with E.
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0002.gif)
where
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0003.gif)
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0004.gif)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0005.gif)
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0006.gif)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0007.gif)
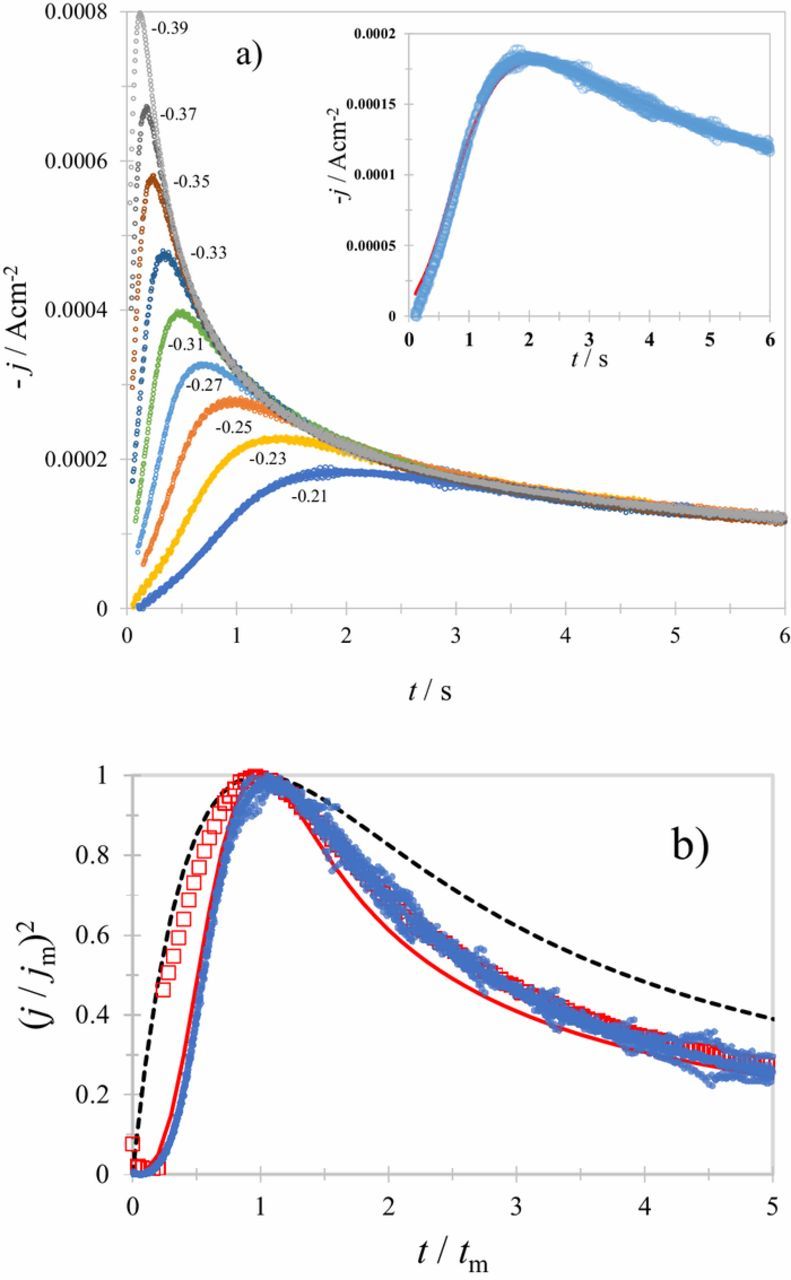
Figure 3. a) Family of potentiostatic current density transients obtained after subtraction of the respective blank from the j-t plots shown in Figure 2. The inset shows the comparison of a theoretical transient (red solid line) obtained after nonlinear fitting of Eq. 8 into the experimental data (points) recorded at η = −0.21 V. The best-fit parameters are shown in Table I. b) Comparison of non-dimensional plots of two of the experimental j-t transients shown in Figure 3a recorded at η = −0.21 (circles) and η = −0.35 V (squares) with the theoretical dimensionless plots for instantaneous (dashed line) and progressive (solid line) nucleation mechanisms (see Scharifker et al.44,45).
Table I. Best-fit parameters and diffusion coefficient obtained from the experimental potentiostatic current transients depicted in Figure 3 using Eq. 8.
| 104 P1/ | P2/ | A/ | 10 8 N0/ | 10 7 D/ | |
|---|---|---|---|---|---|
| −η /V | Acm−2s1/2 | s−1 | s−1 | cm−2 | cm2s−1 |
| 0.21 | 2.69 ± 0.01 | 1.4 ± 0.2 | 0.92 ± 0.02 | 0.5 | 2.44 |
| 0.23 | 2.77 ± 0.01 | 2.1 ± 0.1 | 1.15 ± 0.02 | 0.8 | 2.59 |
| 0.27 | 2.80 ± 0.01 | 4.3 ± 0.2 | 2.51 ± 0.02 | 1.5 | 2.65 |
| 0.31 | 2.87 ± 0.01 | 6.1 ± 0.1 | 3.26 ± 0.02 | 2.1 | 2.78 |
| 0.33 | 2.86 ± 0.01 | 19.1 ± 0.1 | 3.39 ± 0.02 | 6.6 | 2.76 |
| 0.35 | 2.92 ± 0.01 | 26.5 ± 0.1 | 3.69 ± 0.02 | 8.8 | 2.88 |
| 0.37 | 3.01 ± 0.04 | 35.7 ± 0.3 | 4.47 ± 0.04 | 11.1 | 3.06 |
| 0.39 | 3.01 ± 0.04 | 48.5 ± 0.3 | 5.25 ± 0.03 | 15.1 | 3.06 |
In these equations F is the Faraday constant, z is the number of electrons transferred, λ is the integration variable, M and ρ are the Pd atomic mass and density, respectively, F is the Faraday constant, A is the palladium deposit nucleation frequency and N0 is the number density of active sites for palladium nucleation on the electrode surface. The numerical values a = 0.051314213, b = 0.47910725, c = 1.2068142 and d = 1.185724 correspond to the parameters of the polinomial approximation to the Dawson integral.
Therefore, Eqn. 2 can be parameterized as follows:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0008.gif)
Once determined the values of the nucleation frequency A, the analysis of the ln(A) vs. η dependence as described on37 allows determination of the fundamental kinetic parameters α (the transfer coefficient) and j0 (the exchange current density), which in turn permit estimation of the critical nucleus nc size and the Gibbs free energy of nucleation ΔG(nc). The fitting of the ln(A) vs. η values in Table I to Eq. 9 is shown in Figure 4 and results in α = 0.013 and j0 = 0.0012 A cm−2. With these values, the overpotential-dependent values of the thermodynamic quantities ΔG(nc) and nc for the nucleation of Pd from the choline chloride/ethylene glycol DES can be estimated, as summarized on Table II.
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0009.gif)
With
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0010.gif)
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0011.gif)
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/166/1/D3205/revision1/d0012.gif)
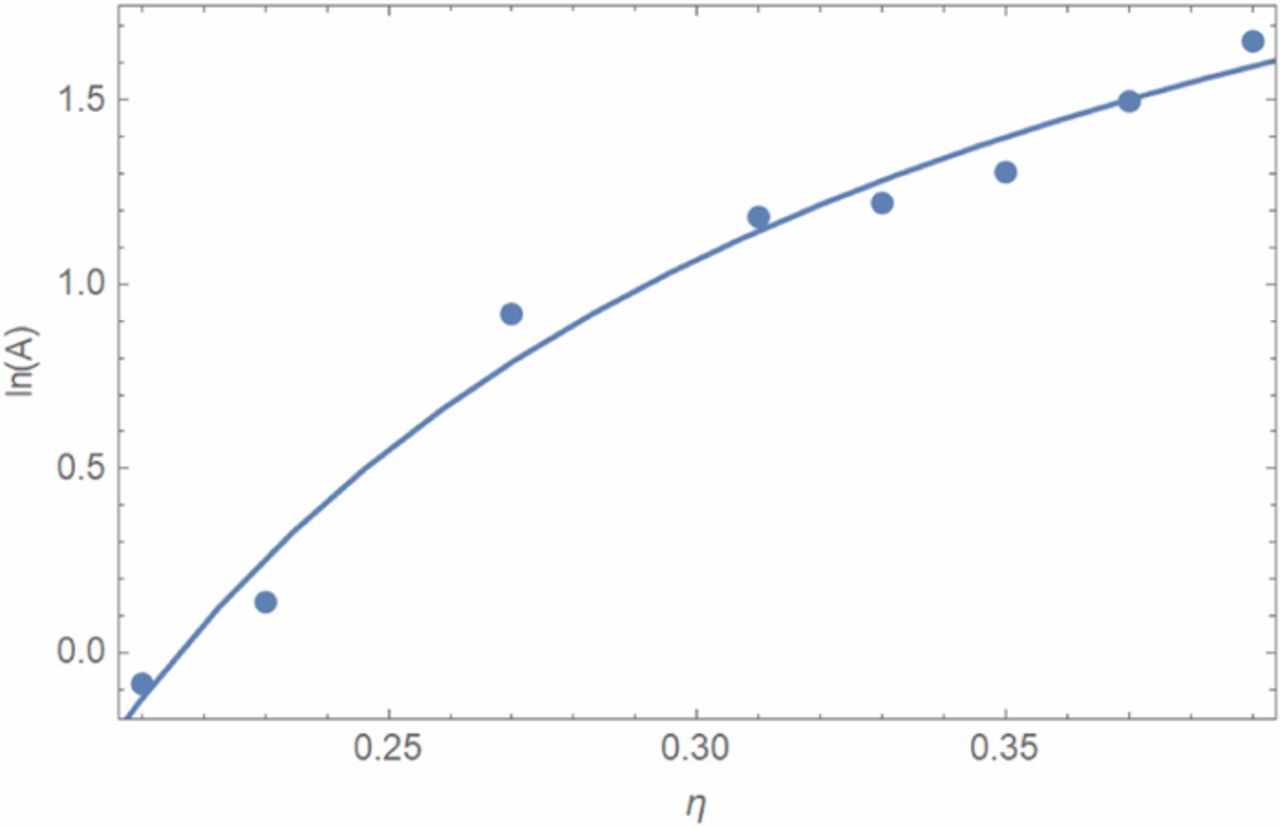
Table II. Thermodynamic parameters for the nucleation of Pd onto GCE from a 1:2 molar ratio choline chloride and ethylene glycol DES at 298 K.
| η / V | ΔG(nc) / J mol−1 | nc |
|---|---|---|
| 0.21 | 5343 | 0.26 |
| 0.23 | 4454 | 0.20 |
| 0.27 | 3232 | 0.12 |
| 0.31 | 2452 | 0.08 |
| 0.33 | 2164 | 0.07 |
| 0.35 | 1924 | 0.06 |
| 0.37 | 1721 | 0.05 |
| 0.39 | 1549 | 0.04 |
Where νa is the atomic volume, e is the elemental charge, z+ is the charge number of the depositing ion, σ is the surface tension, k is the Boltzmann constant and T is the absolute temperature.
As observed before in ref,37 the α values obtained for the deposition of Pd from DES are rather low (0.013) as compared with the values obtained for metal electrodeposition in aqueous media. However, as discussed in37 and references therein, the electrodeposition of metals is quite a different phenomenon from the outer shell electron transfer reaction from which the transfer coefficient α is defined by the Marcus theory. In a media such as DES is reasonable to expect a high complexation of the metallic species, which in turn could determine that the intermediate transition state of the electron transfer reaction is more similar to the metal ion species in solution (or in adsorbed state on the electrode surface) thus, rendering an asymmetric energy barrier for electron transfer. On the other hand, the j0 values obtained indicate some kinetic facility, which correlates with the information provided by the thermodynamic parameters of nucleation ΔG(nc) and nc, i.e., low values for the energy barrier for nucleation and the fact that surface active sites occupied with a reduced single metallic atom are already stable entities for further metal ion aggregation and nucleus growth (nc values < 1).
SEM and XPS analysis
Figure 5 shows SEM images obtained on the GCE surfaces after applying a potentiostatic current transient at η = −0.35 V during 120 s. It is worthy of mention that this overpotential value was chosen because during the PdNPs early stages, the transient follows quite close the instantaneous nucleation mechanism (see Figure 3b), i.e., a fast saturation of available nucleation active sites, a fact consistent with the ΔG(nc) and nc values obtained and also with the empirical information depicted in the SEM images shown in Figure 5. In such conditions a high density of monodisperse small sized nuclei should be expected as effectively observed. Furthermore, from integration of this current density transient, the electric charge, q = 2.6 × 10−3 C, involved during the PdNPs formation was calculated and from this q value and the Faraday's first law of electrolysis, the mass of the deposited palladium was obtained (1.43 × 10−3 mg). Now, regarding these images, it is possible to note that PdNPs formation took place all over the substrate surface, displaying diameters of (41 ± 5) nm, then since the standard deviation is smaller than 15% it can be considered that the PdNPs are monodisperse in size. As can be noted from the size distribution histogram in Figure 4, the PdNPs follow closely a normal distribution. These results support the prediction regarding the instantaneous nature of the Pd deposition at the applied overpotential.
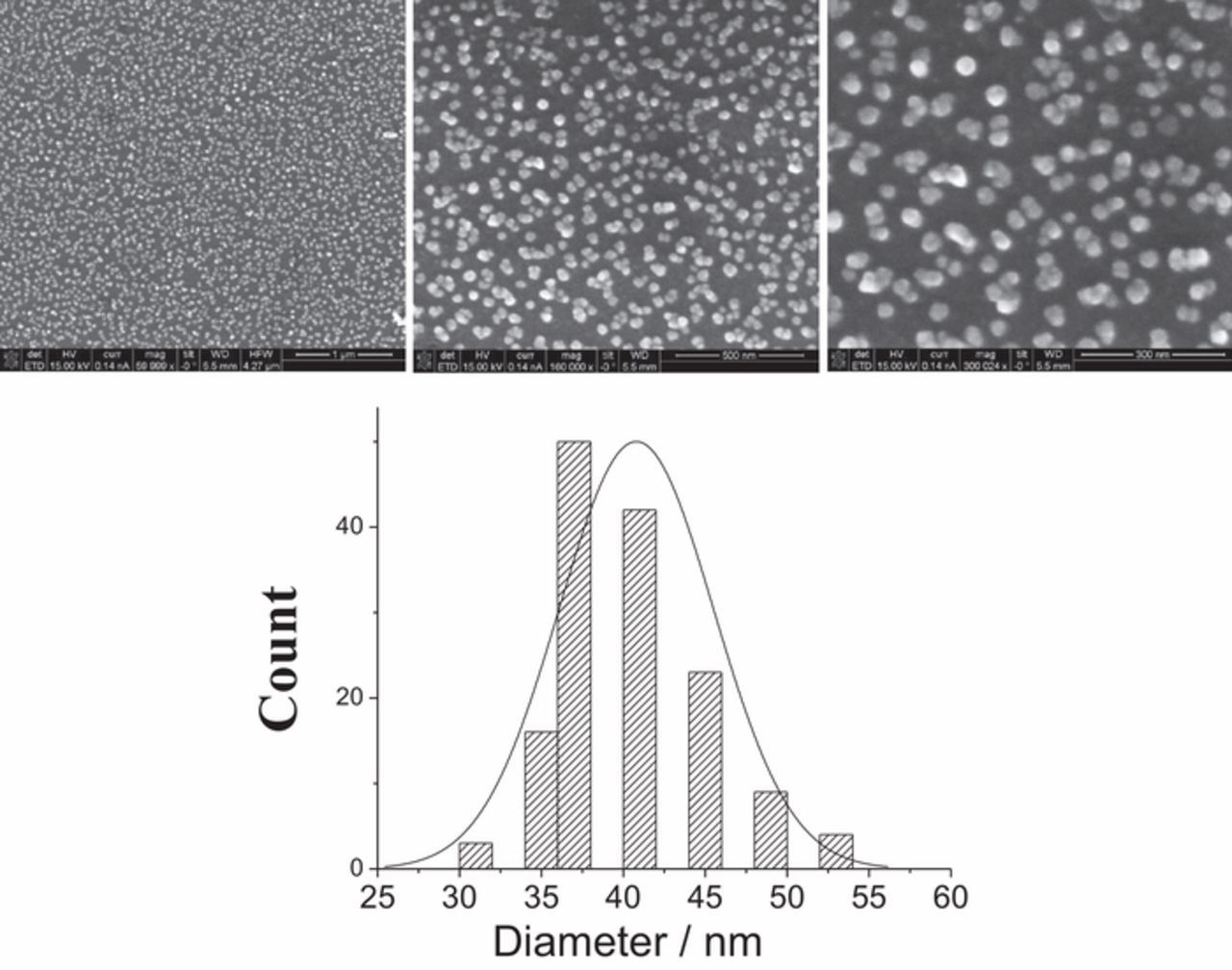
Figure 5. SEM images corresponding to the surface of the GCE electrodeposited with PdNPs at –0.34 V during 120 s., at different magnifications: left) 60,000, center) 160,000 and right) 300,000 X, and the size distribution histogram of the PdNPs estimated from analysis of the SEM images along with the normal distribution curve.
As can be concluded from the XPS analysis, see Figure 6, taken on the surfaces shown in Figure 5, the PdNPs are mainly compounded by metallic Palladium, Pd(0), and a small amount of either, PdO or PdO2. It is important to stress that the XPS spectra in Figure 6 are quite similar to those reported by Shrestha et al.,24 for palladium electrodeposited from a 1-butyl-1-methylpyrrolidinium dicyanamide ionic liquid.
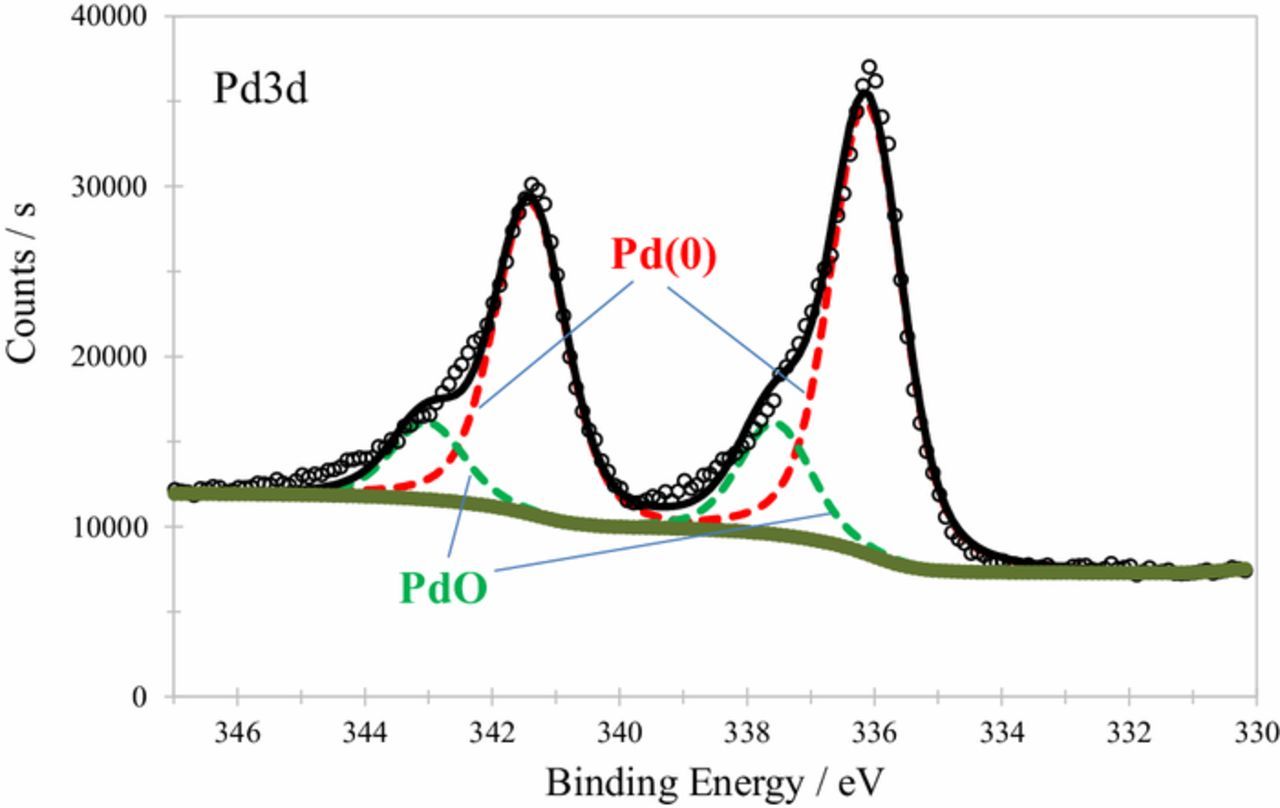
Figure 6. Experimental (○) XPS spectra, of Pd3d region, carried out on the surface of the GCE electrodeposited with PdNPs, same sample as in Figure 4, the solid line is the fitting of XPS profiles multiplex with the sum of Lorentzian and Gaussian functions, using the Avantage 5 software. The Pd3d peak was split into 3d5/2 and 3d3/2 peaks, each of which was fitted with peaks representing metallic (Pd0) and oxidized (Pdm+) states. Some of the Pdm+ could be PdO and PdO2.
Figure 7a depicts a CV recorded in the system GCE/PdNPs/0.1 M HClO4 where the PdNPs were deposited on the GCE as indicated in SEM and XPS analysis section. The typical response due to palladium oxidation to form palladium oxides within the forward potential scan (between 0.55 and 1 V), its corresponding reduction during the backward scan (between 0.6 and 0.4 V) and the associated hydrogen processes region on metallic palladium (between 0.05 and −0.2 V) are clearly present in the CVs. This experimental evidence further supports the presence of PdNPs on the GCE after applying the potential step. Figure 7b shows a CV obtained in this same system but after bubbling CO during 15 minutes, followed by N2 during 20 minutes at a constant potential of −0.2 V. During the first cycle, it is possible to note the formation of a sharp anodic peak (between 0.6 and 0.7 V) due to adsorbed CO striping off the PdNPs. The second cycle depicts the typical Pd response in acid media. From integration of the CO striping peak, the electroactive surface area of the PdNPs was estimated as (1.00 ± 0.05) cm2, around 5 times bigger than the geometric area of the GCE.
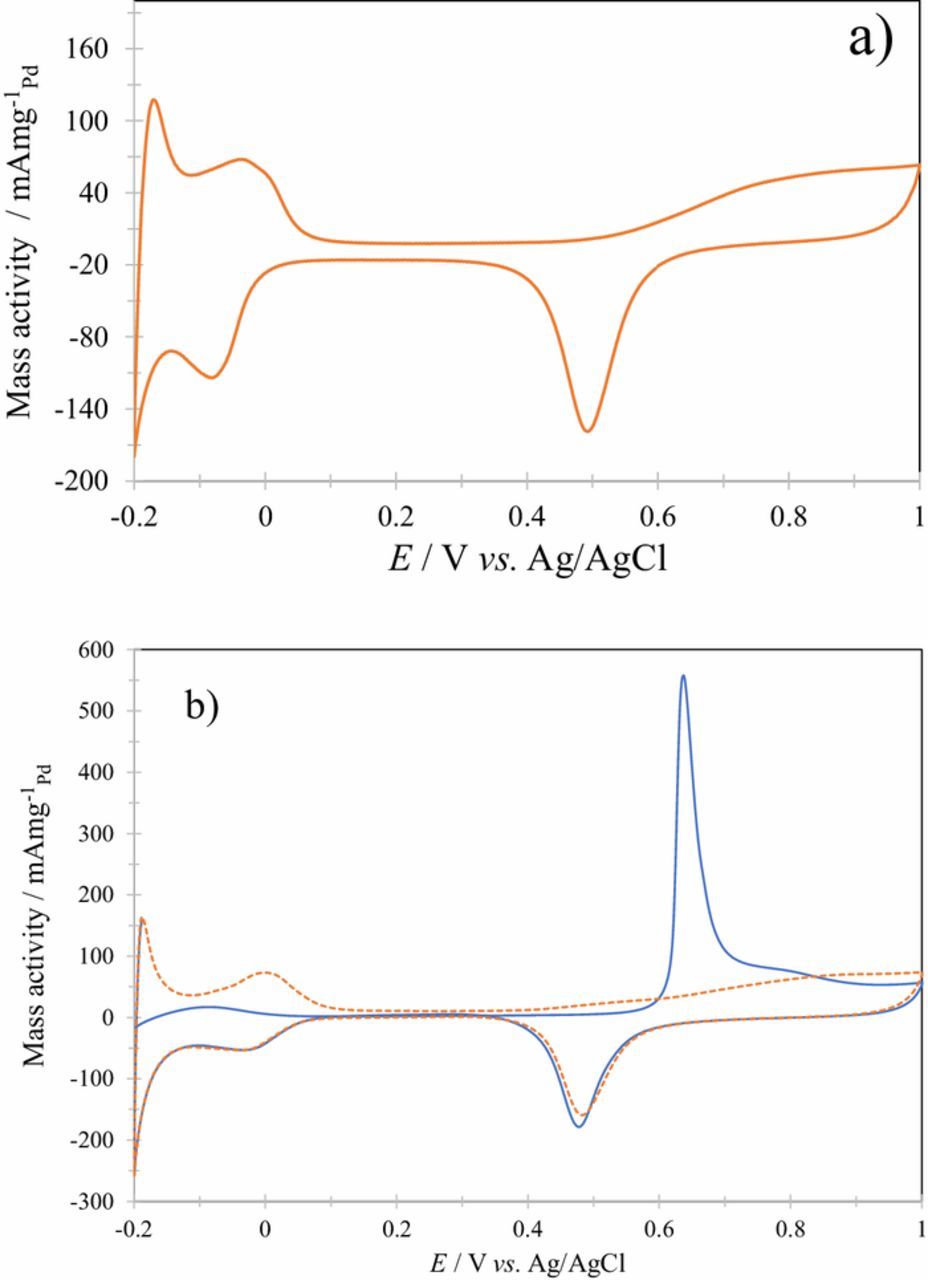
Figure 7. Cyclic voltammograms recorded with the GCE/PdNPs electrode immersed in 0.1 M HClO4 a) at 100 mV s−1 and b) after bubbling CO first 15 minutes and then N2 during 20 minutes (applying a constant potential of −0.2 V) at 20 mV s−1 scan rate, the solid and broken lines correspond to the first and second voltammetric cycles, respectively.
Electrochemical oxidation of formic acid
The catalytic performance of the PdNPs electrodeposited on the GCE surface, as indicated in SEM and XPS analysis section, normalized against the mass of electrodeposited Pd toward the formic acid electrochemical oxidation is presented in Figure 8a. The mass activity reached a maximum, 4200 mAmgPd−1, at around 0.5 V. It is important to note that the poisoning tolerance, which is defined as the peak current ratio of the forward scan to the backward scan,49 which is practically 1.0, strongly suggests that the PdNPs surfaces are not poisoned with formic acid oxidation products. Figure 8b shows chronoamperometries recorded at different applied potentials during formic acid oxidation, using the same electrodes as in Figure 8a. The mass activity transients are characterized by a decrease in the mass activity over time that has been attributed to the poisoning effects of the species formed during dissociative adsorption of formic acid.26 From this figure, it is possible to note that the higher steady state mass activity corresponds to the plot recorded at 0.2 V (ca. 172 mAmgPd−1). Table III depicts a comparison of the electrochemical performance of different PdNPs-based electrodes reported in the literature, toward formic acid oxidation. From this comparison it can be noted that the GCE modified with PdNPs, by means of potentiostatic electrodepositon from the choline chloride and ethylene glycol deep eutectic solvent at 298 K, performs an outstanding activity.
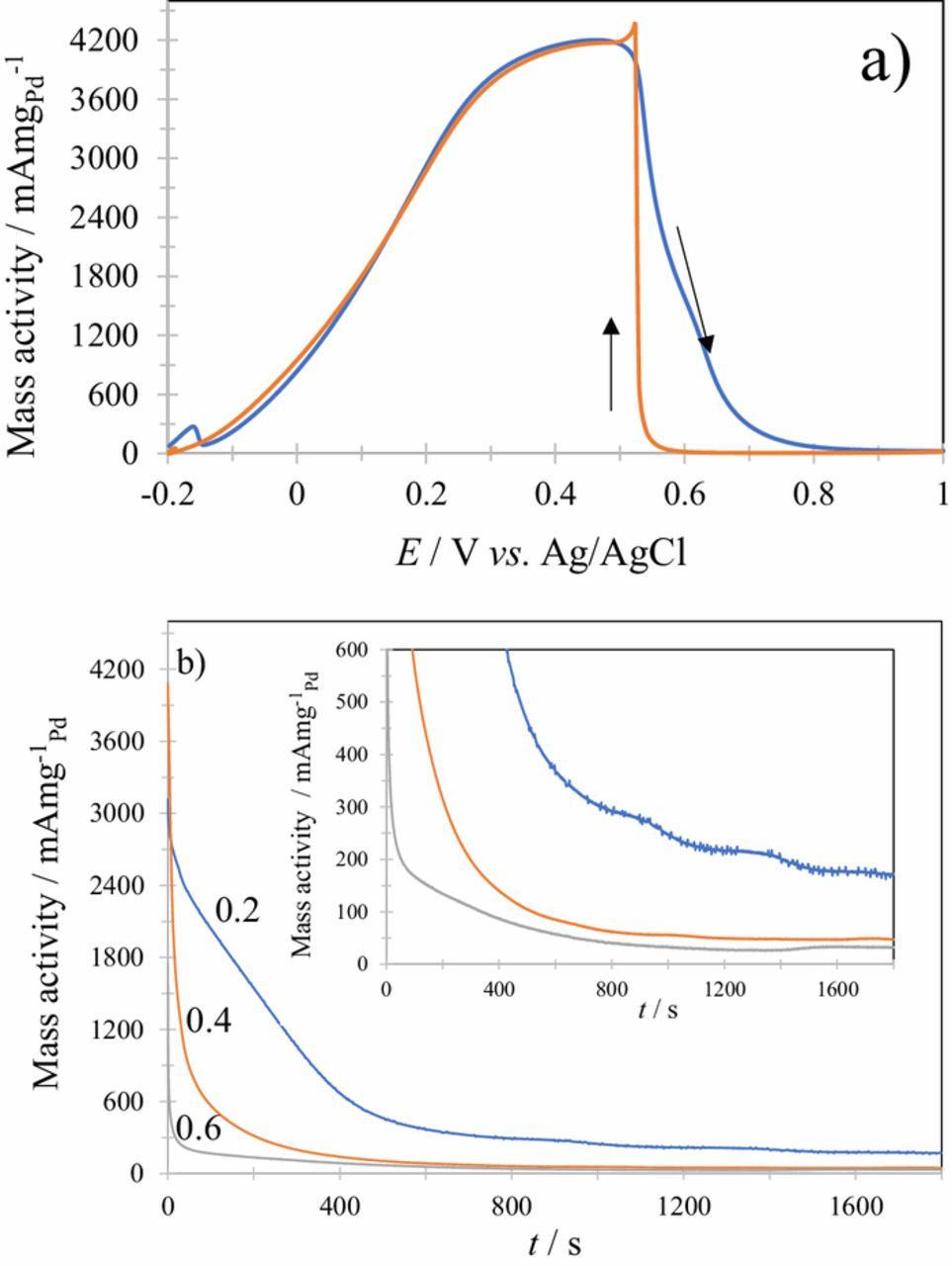
Figure 8. a) Cyclic voltammogram recorded in the system GCE/PdNPs/1.0 M HCOOH, 0.1 M HClO4 at 20 mVs−1 potential scan rate, starting at −0.2 mV in the positive direction and switching the potential at 1 V. b) Chronoamperometric curves of PdNPs/GCE for formic acid oxidation, polarized at different constant potentials, indicated in the figures in V. The inset shows a close up of the mass activity.
Table III. Comparison of the mass activity (mAmgPd −1) of different electrodes toward the FAOR.
| Electrode | NPs synthesis method | Potentiodynamic* | Potentiostatic*,** | Reference |
|---|---|---|---|---|
| GCE/NPs | Potentiostatic electrodeposition from DES | 4200 ± 100 | 172 ± 2 | This work |
| C/PdNPs | Moderate wet chemistry method | 250 | 50 | 9 |
| C/CuPdAuNPs | Moderate wet chemistry method | 550 | 50 | 9 |
| C/PdCoNPs | Simultaneous reduction reaction using NaBH4 | 250 | 50 | 12 |
| C/PdNPs | reduction reaction using NaBH4 | 150 | 20 | 12 |
| C/PdNi | Sonochemical reactions | 2500 | - | 15 |
| C/PdCo | Sonochemical reactions | 2250 | - | 15 |
| C/PdMn | Sonochemical reactions | 2000 | - | 15 |
| C/PdFe | Sonochemical reactions | 2490 | - | 15 |
| C/Pd | Sonochemical reactions | 500 | - | 15 |
| C/Pd | reduction reaction | 500 | 100 | 16 |
| MWCNTs/Pd | reduction reaction | 600 | 90 | 16 |
| Carbon cloth /Pd/PdCoNPs | Potentiostatic electrodeposition from aqueous solution | 1580 | 400 | 26 |
| Carbon cloth /PdNPs | Potentiostatic electrodeposition from aqueous solution | 540 | 100 | 26 |
Conclusions
It has been shown from analysis of potentiostatic current density transients recorded during palladium electrodeposition onto GCE from the choline chloride and ethylene glycol deep eutectic solvent at 298 K, that the formation of PdNPs occurs via 3D nucleation with diffusion-controlled growth (3D-DCG). The analysis of the current transients via available 3D-DCG formalisms provides values for the kinetic and thermodynamic fundamental parameters of the nucleation of Pd onto GCE from DES consistent with the observed empirical evidence provided by SEM images. Furthermore, it was also shown that the PdNPs, formed mainly by metallic palladium with small amounts of palladium oxides, exhibited an outstanding electrochemical performance toward the formic acid oxidation, which is even better than other PdNPs-based electrodes reported in the literature.
Acknowledgments
The authors like to thank CONACyT for project 258487. IEEL thanks CONACyT for the support given to undertake postgraduate studies at UAMA and a research stay in the Universitat de Barcelona under the supervision of Prof. Elvira Gómez. MGMY, MTRS, MRR and MPP thank the SNI for the distinction of their membership and the stipend received.
ORCID
M. Romero-Romo 0000-0002-5783-5776
J. Mostany 0000-0002-3163-4001
M. Palomar-Pardavé 0000-0002-2944-3599